結合型D-アミノ酸についての計算機的研究

小田 彰史
従来、タンパク質を構築するアミノ酸残基はL-体のみであると考えられてきた。しかし近年タンパク質中に残基として含まれるD-アミノ酸が発見されており、それが白内障やアルツハイマー型認知症など、加齢性変化と関連している可能性があることが指摘されている(1)。このような、残基としてタンパク質中に含まれるD-アミノ酸を結合型D-アミノ酸と呼ぶが、この結合型D-アミノ酸はこれまでに水晶体中のクリスタリンや脳内のβアミロイドなどから検出されている。これまでに検出されている結合型D-アミノ酸は主にアスパラギン酸 (Asp) であるが、セリン残基の反転についても観測されている。AspについてはD-α-Aspだけではなく、異性化したL-β-AspおよびD-β-Aspも検出されており、野生型のL-α-Aspとあわせて4種類の異性体が存在していることになる。
これら結合型D-アミノ酸についての研究は未だ発展中の段階にあり、解明すべき問題が多数残されている。タンパク質中でD-アミノ酸残基がどのように生じたのか、またD-アミノ酸残基が生じることによって何が起こるのか、そしてD-アミノ酸残基は体内でどのように処理されていくのか、その全てを解明する必要がある。すなわちタンパク質中での結合型D-アミノ酸の一生について、「D-アミノ酸はどこから来たのか、D-アミノ酸は何者か、D-アミノ酸はどこへ行くのか」を取り扱わなければならない。このような場合、実際に実験を行うことなく、コンピュータによって様々な状況を再現しうる計算化学技術は有力な研究手段となり得る。
自然科学の研究は、従来実験と理論から成り立っていた。実験で得られたデータから普遍的な理論を導き出し、また理論から期待できる現象を実験によって実現するというサイクルで、自然科学は発達してきた。しかし科学の進歩に伴って実験・理論ともに高度に複雑化し、人間が扱うにはあまりにも多すぎるデータの得られる実験や、人間には解釈困難なほど多数の数式を駆使した理論なども珍しくなくなった。その結果、自然科学を扱うための第三の手法として計算の果たす役割が大きくなってきた。すなわち、大量の実験データを計算で処理することで普遍的な傾向を見出したり、複雑な理論を元に数値計算を行うことで現実に起こりうる現象を予測したりといった、理論と実験とを円滑につなぐための役割を計算は果たしている(図1)。20世紀後半以降のコンピュータの爆発的な進歩も駆動力となり、自然科学のための計算技術は近年大きく発展しつつある。
本稿では、結合型D-アミノ酸を研究するに当たって計算化学手法がどのように利用可能であるか、筆者らの研究成果を交えて概説する。
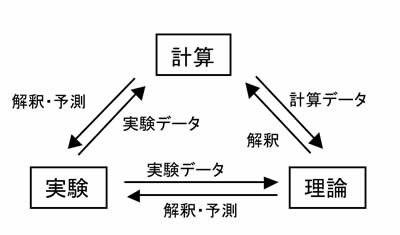
図1 自然科学を扱うための3つの手法。実験で得られたデータを理論で解釈したり、新たな現象を予測したりする。計算はその橋渡しとして、データの整理や解釈の手助けを行う。
計算化学手法は大きく分けて2つに分類することができる。1つは量子化学計算手法で、これは量子力学を用いて分子・原子の性質を調べるための手法である。Schrödinger方程式から得られる波動関数を用いることによってあらゆる観測可能量を求めることが可能であるため、実験を一切行うことなく量子化学計算のみによって分子・原子の様々な性質を算出することができる。また運動量の小さい物体ほど量子論効果が無視できないため、分子・原子を構成する要素のうち電子の性質を求めるには量子化学計算が必須となる。
もう1つの計算化学手法は分子力学法で、これは古典力学に基づいた計算手法である。すなわち、分子や原子をあたかも通常の(古典力学に従う)物体であるかのように扱って計算を行う。分子力学法では、分子の構造変化がエネルギーに与える影響についてパラメータ化した分子力場を用い、特定の構造の分子がどのようなエネルギーをもつのかを簡便に計算する。従って高速な計算が可能であり、生体高分子のような比較的大きな分子に対しても容易にエネルギーを算出することができる。
量子化学計算と分子力学法には、それぞれ長所と短所が存在する。量子化学計算は上述の通り様々な物理量を求めることが可能で、かつ電子も扱うことができる。一方で分子・原子に対するSchrödinger方程式を厳密に解くことはほぼ不可能であり、近似を導入したとしても莫大な量の計算コストを要する。生体高分子の量子化学計算を行うための工夫も開発されてはいるものの、溶媒効果の導入や経時変化の観測などには困難を伴うことが多い。一方の分子力学法では迅速な計算が可能であり、生体内を模した環境中での分子の挙動をシミュレートすることなども可能であるが、一方で古典力学に基づいた手法であるために電子を扱うことが事実上不可能である。分子・原子の性質の多くが電子によって決まるため、分子力学法ではそれら性質を求めることができない。例えば化学反応は分子・原子における電子の移動を伴う現象であるが、分子力学法で化学反応を扱うのは困難である。このようにそれぞれに適用できる限界が存在するため、目的に応じて手法を使い分ける必要がある。
計算化学手法にはこの他にも情報学的な技術を化学に適用したケモインフォマティクス手法も存在し、例えば定量的構造活性相関やdrug-likenessなど、創薬の分野で利用されている技術などが含まれる。
3.1. D-アミノ酸はどこから来たのか
タンパク質中の結合型D-アミノ酸の起源としては、現在主に非酵素的反応によって生じる機構が調べられている。特に多くの立体反転が観測されているAsp残基については図2のような機構が提唱されている(1)。この機構ではAsp残基の側鎖のカルボキシル基がC端側隣接残基のアミド窒素と結合を形成し、5員環のスクシンイミド(Suc)構造となる。Suc構造はケト-エノール互変異性によってエノール型となるが、これによってAsp残基の不斉炭素がキラルではなくなる。このエノール型がケト型に戻る際、炭素がキラルではなくなっているためにL-体だけではなくD-体のSucが得られることとなる。また、環が開裂する際にどちらのアミド結合が切断されるかによってα-Aspとβ-Aspとが生じる。
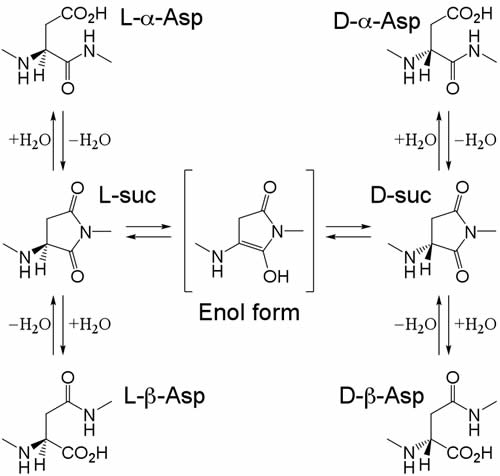
図2 アスパラギン酸の異性化機構
アミノ酸残基の立体反転は化学反応であり、これを計算によって直接再現しうるのは量子化学計算のみである。量子化学計算手法には分子軌道法、原子価結合法、密度汎関数法(以下DFT法)があるが、我々の研究では近年広く使用されているDFT法を用いた(2, 3)。DFT法は波動関数を電子密度の汎関数として表現する方法で、比較的高速に高精度な結果が得られる手法として有機物・無機物問わず様々な物質の計算に用いられている。
Asp残基が反転する際に、最初に鍵となるのは側鎖の環化である。この反応がどのように進行するかを解明することで、Asp残基が反転するために必要な条件を求めることができる。この反応はアミド窒素によるカルボキシル基の炭素への求核置換反応として記述される。しかし一般にアミド窒素の求核性は高くないことが知られており、実際に量子化学計算によって求められた活性化障壁も実験結果と比較して高い値となっている。そこで我々は、アミドが互変異性化によってイミノールとなり、イミノールの窒素が求核攻撃を行う機構を考察した(図3)(2, 3)。溶媒の水分子が触媒として機能するモデルを作成してDFT法によって計算を行ったところ、活性化障壁が30 kcal/mol以下と実験データをよく再現する値となった。このことから、Asp残基の反転にはイミノール体を経由する可能性があること、また水分子が触媒として働きやすい残基が立体反転を起こしやすいことが示唆された。
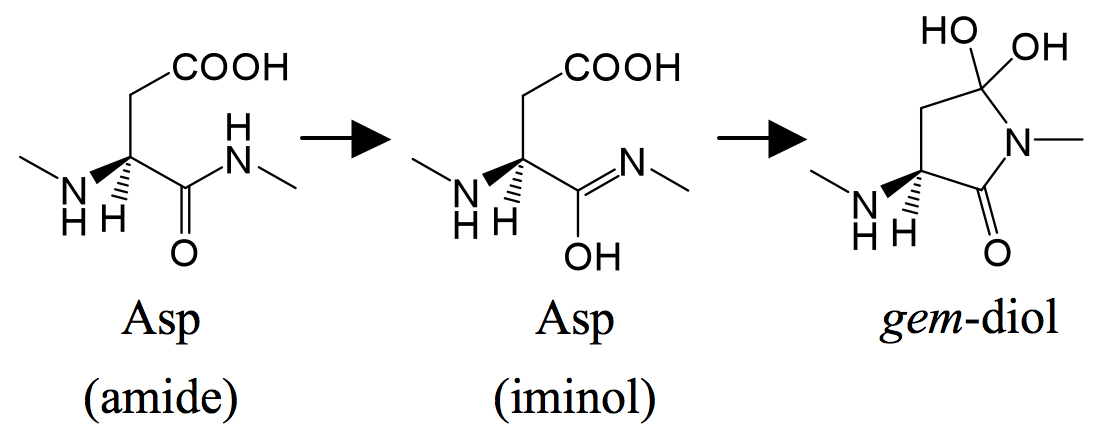
図3 イミノール型構造を経由した環化反応。アミドがイミノールとなることで求核性が増強され、四面体型中間体であるgem-ジオール構造へと変化しやすくなる。
3.2. D-アミノ酸は何者か
タンパク質中のアミノ酸がD-体となることで、そのアミノ酸周辺の環境が劇的に変化する。例えばAsp残基の反転であれば、カルボキシル基を含む側鎖がα水素と入れ替わることを意味するため、かさ高さも周囲の静電ポテンシャルも大きく影響を受ける。この変化はタンパク質全体の立体構造に影響を与え、タンパク質の変性を引き起こすことがある。また、系によってはタンパク質の凝集へとつながることも考えられる。このようにタンパク質中のアミノ酸残基の立体反転は、タンパク質の構造・物性に影響を与える。
タンパク質中のD-アミノ酸残基によって何が引き起こされるかについて計算するには、分子力学法による計算が適している。アミノ酸残基の反転の影響は局所的なものに留まらず、タンパク質全体、ひいては生物個体の老化とも関係している可能性が示唆されており、周辺環境も含めたタンパク質全体の計算を行うことが望ましい。量子化学計算でタンパク質を扱う際にはタンパク質の一部分のみをモデル化したり、溶媒の効果を取り入れない、あるいは簡単にしか取り入れなかったりすることがほとんどであるため、大きな系の全体構造を計算するには不適当である。アミノ酸残基の立体反転が構造に与える影響を詳しく調べるため、我々は分子動力学(MD)シミュレーションを行った(4, 5)。MDシミュレーションはニュートンの運動方程式に基づいて系の挙動をシミュレートする手法であるが、これを用いることで300 Kの条件下でのタンパク質の構造変化を再現することが可能となる。
Asp残基の立体反転が見られるタンパク質・ペプチドはいくつかあるが、その中で比較的よく調べられており、かつ短いため計算の容易な系としてβアミロイド(Aβ)が挙げられる。Aβの中でもアルツハイマー型認知症と強く関連していると考えられているAβ 1-42には、配列中に3つのAsp残基が存在する(Asp1, Asp7, Asp23)。Sakai-Katoらはこの3つのAsp残基の全ての組み合わせについて網羅的に立体反転を起こしたペプチドを合成し、その二次構造および凝集性について調べた(6)。Aβ1-42は通常ランダムコイルの状態であるが、Sakai-Katoらの研究によるとAsp23の立体反転によってAβ1-42がβ構造をとりやすくなり、凝集しやすくなると報告されている。そこで我々はAsp23の立体反転したAβ1-42をコンピュータ上で作成し、その立体構造を調査した。
Aβ1-42のとり得る配座を詳細に再現するため、計算にはレプリカ交換MD(REMD)法(7)を用いた。これは高温下でのシミュレーションの効果を取り込み、構造変化を効果的に促進することのできるシミュレーション手法である。269.7 Kから600.0 Kまでの16通りの温度を用いて計算を行ったが、解析には300 Kの結果を用いた。分子力場としてはAMBER力場のff99SB力場を用いた。タイムステップは2 fs、全部で50 nsのシミュレーションを行い、構造は1 psごとに抽出した。溶媒環境としては一般化Born法のGBOBC法を用いた。REMDの計算にはAMBER10(8)を、計算の準備および結果の解析にはAmberToolsを用いた。
REMDシミュレーションの結果を用いてペプチドの二次構造を評価してみると、全てL-アミノ酸からなるペプチドではβ含量が25.8%だったのに対してAsp23を立体反転させたペプチドでは34.5%となっており、実験データの示す通りAsp23の立体反転によってβ構造をとりやすくなっていることがわかる。また、図4に示した通りAβ1-42のN端側、C端側どちらにもβ構造が現れていた。これらの結果は単量体のAβ1-42に対するシミュレーションによって得られており、Asp23の立体反転がAβ1-42単独での配座に影響を与えていることを示している。Aβ1-42の凝集のメカニズムとして、まずAβ1-42のC端側のヘリックス同士が引き合い、複数のAβ1-42が集まった後にβ構造へと変化する機構が提唱されている(9)。その結果さらに多くのAβ1-42が凝集し、沈殿へとつながる。しかしこの計算の結果から、Asp23を立体反転させると単量体のまま(凝集する以前に)β構造をとりやすくなる可能性が示唆された。すなわち、これらD-Aspを含むAβ1-42は野生型とは異なったメカニズムで凝集する可能性があると考えられる。
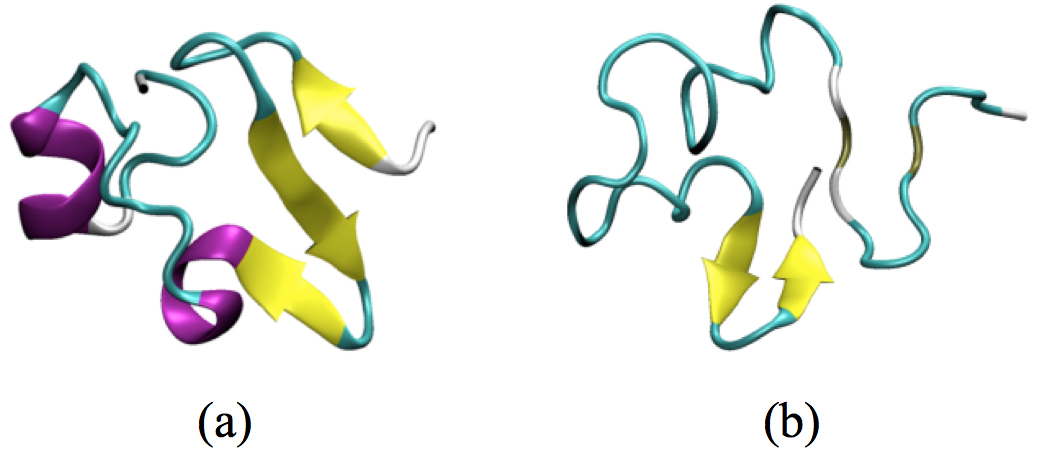
図4 β構造を含む配座の例。
(a) N端側にシートがある構造(38 ns時点)
(b) C端側にシートがある構造(22 ns時点)。
3.3. D-アミノ酸はどこへ行くのか
図2に示したように、Asp残基には全部で4種類の異性体が存在する。しかしながら生体内では主にL-α-AspとD-β-Aspの検出量が多く、D-α-AspとL-β-Aspについては比較的少ない量しか検出されていない(1)。これを説明する理由の1つとしてD-α-AspとL-β-Aspを認識してL-α-Aspへと修復する反応が起こっているのではないかという説が提案されている。そのような反応に関与する酵素として、タンパク質L-イソアスパラギン酸/D-アスパラギン酸メチルトランスフェラーゼ(PIMT)が挙げられることがある。
PIMTはタンパク質・ペプチド中のD-α-Asp残基とL-β-Asp残基を認識し、その側鎖のカルボキシル基をメチルエステル化する酵素である(図5)(10)。メチルエステル化した側鎖は求核攻撃を受けやすくなり、環構造をとりやすくなる。スクシンイミド構造となった後は図2の反応が進行するため、4種のAspのいずれにもなる可能性がある。すなわち、PIMTによってD-α-AspとL-β-Aspだけが中間体へと変化し、4種いずれかのAspになるため、結果としてD-α-AspとL-β-Aspが消費されて減少していくことになる。
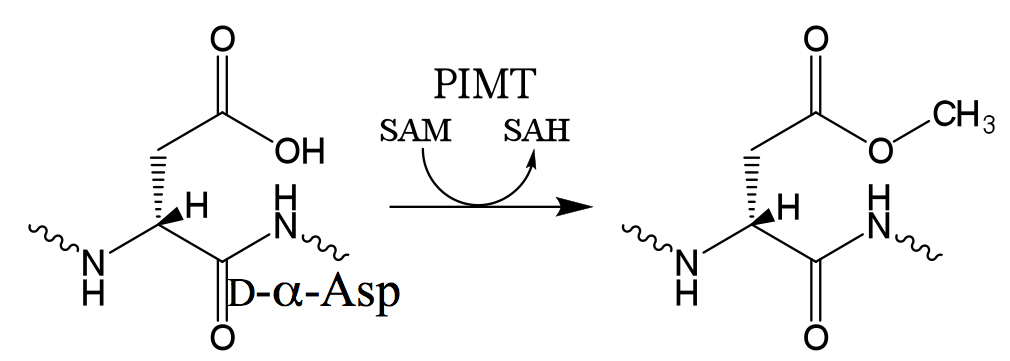
図5 D-α-Asp残基に対するPIMTの働き
我々はヒトPIMTが基質を認識する機構についてコンピュータを用いて検討を行った(11)。その際にはコンピュータによるドッキングでPIMT-基質複合体構造を推定し、さらにMDシミュレーションによって構造緩和を行った。コンピュータによるドッキングはタンパク質とリガンドとの間の相互作用を様々な方法で見積もり、複合体構造や結合親和性を予測する。この際には分子力学法の概念を適用することが多い。我々の研究ではPIMTの基質として6残基ペプチドVYPDHAを用いた。この4番目のAsp残基に、L-α-Asp、D-α-Asp、L-β-Asp、D-β-Aspのそれぞれを用いた4種類のペプチドに対して複合体構造を推定した。このうちD-α-AspおよびL-β-Aspを含むペプチドが実際のPIMTの基質であり、L-α-AspおよびD-β-Aspを含むペプチドは基質ではない。この両者に差があるかどうかを比較した。ドッキングにはLibdock(12)を、MDシミュレーションにはAMBER9を用いた。MDの際の力場にはタンパク質にはff03力場を、リガンドにはGAFFを使用した。タイムステップは2 fs、全体で20 nsのシミュレーションを行った。溶媒の水分子としてTIP3Pモデルを用いてあらわに扱った。
計算の結果、VYP-L-β-Asp-HAペプチドはPIMTと多数の水素結合を形成することがわかった。VYP-D-β-Asp-HAにおいてはVYP-L-β-Asp-HAと類似した位置および配向でPIMTと結合していたにもかかわらず、いくつかの水素結合が形成されなかった。これはVYP-D-β-Asp-HAではペプチドの主鎖にねじれが生じているためではないかと考えられる。また、PIMTとVYP-L-β-Asp-HAとの複合体構造に対するMDシミュレーションの結果、基質の結合していない状態と比べてPIMTのPro50のCγとIle212のCγ2が接近することが分かった。この2つの残基はペプチド基質の中央からC端側付近に近接しており、ペプチド基質を挟み込むような動きをしていた。このようなinduced fit様のタンパク質の動きは他の複合体構造でも観察されており、比較的大きなペプチド基質を認識するために酵素がどのように構造を変化させているかを推測することができる。
本稿では、結合型D-アミノ酸について計算化学手法を用いて解析を行った研究について紹介した。最初に述べたように結合型D-アミノ酸に関する研究は現在進展中であり、そのような分野においては実際に実験を行うことなく様々な条件下での系の性質を調べることのできる計算技術が有効に働く。またタンパク質中のアミノ酸残基が立体反転を起こすことで凝集してしまう場合など、構造生物学的実験が困難な系に対しても、計算化学手法を用いることでタンパク質の立体構造について検討することが可能となる。このようにして得られた成果をさらに実験へとフィードバックし、理論・実験・計算の各分野が協同して研究を進めることで、結合型D-アミノ酸について様々な知見を得ることが可能になると期待している。
文 献
1. Fujii N, Saito T. 2004. Homochirality and life. Chem Rec 4:267-278.
2. Takahashi O, Oda A. 2012. Amide-iminol tautomerization of the C-terminal peptide groups of aspartic acid residues: two-water-assisted mechanism, cyclization from the iminol tautomer leading to the tetrahedral intermediate of succinimide formation, and implication to peptide group hydrogen exchange. In Tyrosine and Aspartic Acid: Properties, Sources and Health Benefits ed. JE Jones, DE Morano, pp. 131-147. Hauppauge, NY: Nova Science Publishers, Inc.
3. Takahashi O. Just three water molecules can trigger the undesired nonenzymatic reactions of aspartic acid residues: new insight from a quantum-chemical study. J Phys Conf Ser, in press.
4. Oda A, Kobayashi K, Takahashi O. 2010. Molecular-dynamics simulations for amyloid β1-42 monomer with D-aspartic acid residues using continuous solvent. Chem Biodivers 7:1357-1363.
5. Oda A, Kobayashi K, Takahashi O. 2011. Comparison of molecular dynamics simulation methods for amyloid β1-42 monomers containing D-aspartic acid residues for predicting retention times in chromatography. J Chromatogr B 873:3337-3343.
6. Sakai-Kato K., Naito M., Utsunomiya-Tate N. 2007. Racemization of the amyloidal β Asp1 residue blocks the acceleration of fibril formation caused by racemization of the Asp23 residue. Biochem Biophys Res Commun 364:464-469.
7. Sugita Y, Okamoto Y. 1999. Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett 314: 141–151.
8. Case DA, et al. 2008. AMBER10, San Francisco, CA: University of California.
9. Kirkitadze M, Condron MM, Teplow DB. 2001. Identification and characterization of key kinetic intermediates in amyloid β-protein fibrillogenesis. J Mol Biol 312:1103-1119.
10. Furuchi T, Homma H. 2007. The role of isomerized protein repair enzyme, PIMT, in cellular functions. Yakugaku Zasshi 127:1927-1936.
11. Noji I, Oda A, Kobayashi K, Takahashi O. 2011. Computational investigation of the substrate recognition mechanism of protein D-aspartyl (L-isoaspartyl) O-methyltransferase by docking and molecular dynamics simulation studies and application to interpret size exclusion chromatography data. J Chromatogr B 873:3310-3316.
12. Diller DJ, Merz KM. 2001. High throughput docking for library design and library prioritization. Proteins: Struct Funct Genet 43:113-124.

小田 彰史(おだ あきふみ)氏
略 歴
1991-1995 大阪大学理学部化学科
1995-1997 大阪大学大学院理学研究科無機及び物理化学専攻博士前期課程
1997-2000 大阪大学大学院理学研究科化学専攻博士後期課程
2000-2006 富山化学工業株式会社
2006-2007 東北薬科大学薬学部助手
2007-2011 東北薬科大学薬学部助教
2007-2008 大阪大学蛋白質研究所特任助教(兼任)
2008-現在 大阪大学蛋白質研究所共同研究員(兼任)
2011-2012 東北薬科大学薬学部講師
2012-現在 金沢大学医薬保健研究域薬学系准教授